Karolina's Bioinformatics Portfolio
View the Project on GitHub akn006-navarro/bimm143_github_redo
Class 07: Machine Learning 1
Karolina Navarro (PID: A19106745)
- Background
- K-means clustering
- Hierarchial clustering
- Principal Component Analysis (PCA)
- PCA of UK food data
- Using base R
Background
Today we will begin our exploration of important machine learning methods with a focus on clustering and dimensionality reduction.
To start testing these methods let’s make up some sample data to cluster where we know what the answer should be.
hist(rnorm(3000, mean = 10))
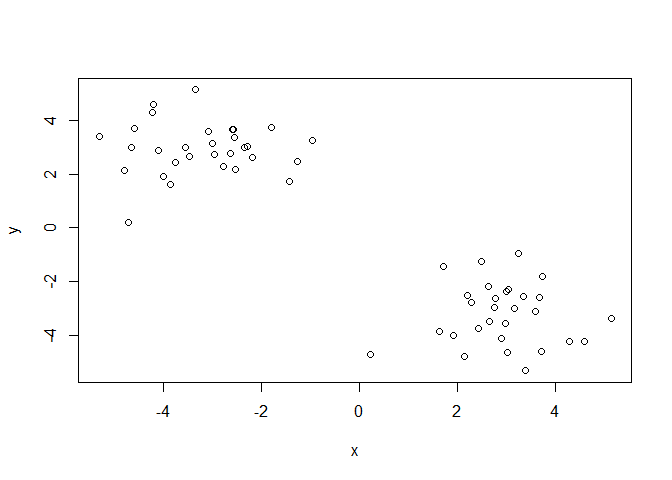
Q. Can you generate 30 numbers centered at +3 and 30 numbers at -3 taken at random from a normal distribution?
tmp <- c(rnorm(30, mean = 3),
rnorm(30, mean = -3) )
x <- cbind(x=tmp, y=rev(tmp))
plot(x)
K-means clustering
The main function in “base R” for K-means clustering is kmeans(), lets
try it out:
k <- kmeans(x, centers = 2)
Q. What component of your kmeans result object has the cluster centers?
k$centers
x y
1 2.941089 -3.185395
2 -3.185395 2.941089
Q. What component of your kmeans result object has the cluster size (i.e how many points are in each cluster)?
k$size
[1] 30 30
Q. What component of your kmeans result object has the cluster membership vector (i.e the main clustering result: which points are in which cluster)?
k$cluster
[1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2
[39] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
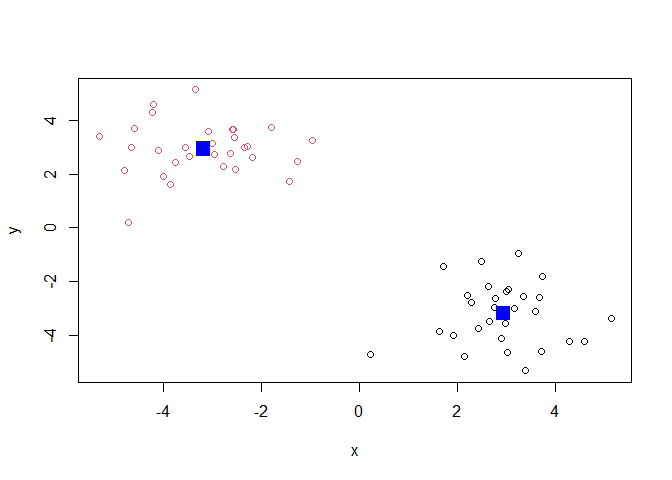
Q. Plot the results of clustering (i.e our data colored by the clustering result) along with the cluster centers.
plot(x, col=k$cluster)
points(k$centers, col="blue", pch=15, cex=2)
Q. Can you run
kmeans()again and clusterxinto 4 clusters and plot the results just like we did above with coloring by cluster and the cluster centers shown in blue?
k4 <- kmeans(x, centers = 4)
plot(x, col=k4$cluster)
points(k4$centers, col="blue", pch=15, cex=2)
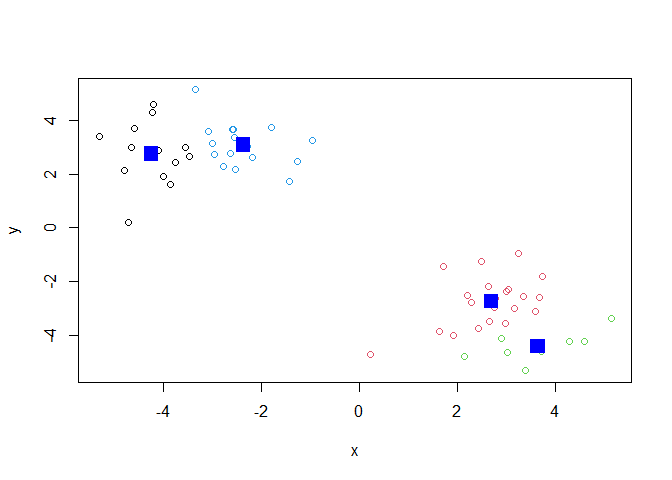
Key point: Kmeans will always return the clustering that we ask for (this is the “K” or “centers” in K-means)!
Hierarchial clustering
The main function for hierarchical clustering in base R is called
hclust(). One of the main differences with respect to the kmeans()
function is that you can not just pass your input data directly to
hclust(). It needs a “distance matrix” as input. We can get this from
lots of places including the dist() function.
Call: hclust(d=d)
Cluster method: Complete Distance: Euclidean Number of projects: 60
d <- dist(x)
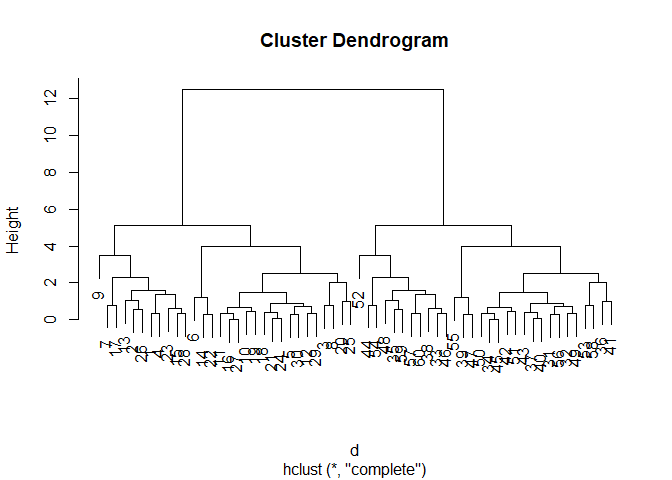
hc <- hclust(d)
plot(hc)
We can “cut” the dendogram or “tree” at a given height to yield our
“clusters”. For this we use the function cutree().
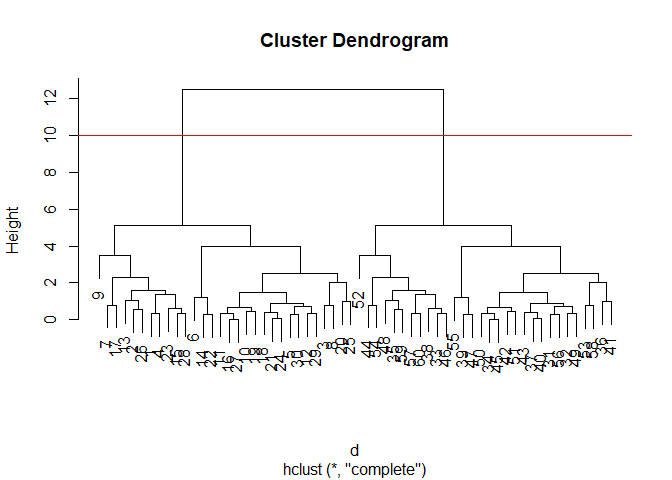
plot(hc)
abline(h=10, col="red")
grps <- cutree(hc, h=10)
grps
[1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2
[39] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
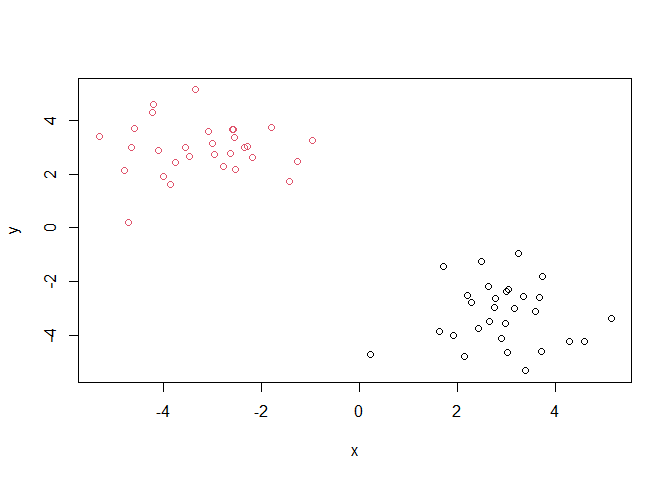
Q. Plot our data
xcolored by the clustering result ftomhclust()andcutree()?
plot(x, col=grps)
Principal Component Analysis (PCA)
PCA is a popular dimensionality reduction technique that is widely used in bioinformatics.
PCA of UK food data
Read data on food consumption in the UK
url <- "https://tinyurl.com/UK-foods"
x <- read.csv(url)
x
X England Wales Scotland N.Ireland
1 Cheese 105 103 103 66
2 Carcass_meat 245 227 242 267
3 Other_meat 685 803 750 586
4 Fish 147 160 122 93
5 Fats_and_oils 193 235 184 209
6 Sugars 156 175 147 139
7 Fresh_potatoes 720 874 566 1033
8 Fresh_Veg 253 265 171 143
9 Other_Veg 488 570 418 355
10 Processed_potatoes 198 203 220 187
11 Processed_Veg 360 365 337 334
12 Fresh_fruit 1102 1137 957 674
13 Cereals 1472 1582 1462 1494
14 Beverages 57 73 53 47
15 Soft_drinks 1374 1256 1572 1506
16 Alcoholic_drinks 375 475 458 135
17 Confectionery 54 64 62 41
It looks like the row names are not set properly. We can fix this
rownames(x) <- x[,1]
x <- x[,-1]
x
England Wales Scotland N.Ireland
Cheese 105 103 103 66
Carcass_meat 245 227 242 267
Other_meat 685 803 750 586
Fish 147 160 122 93
Fats_and_oils 193 235 184 209
Sugars 156 175 147 139
Fresh_potatoes 720 874 566 1033
Fresh_Veg 253 265 171 143
Other_Veg 488 570 418 355
Processed_potatoes 198 203 220 187
Processed_Veg 360 365 337 334
Fresh_fruit 1102 1137 957 674
Cereals 1472 1582 1462 1494
Beverages 57 73 53 47
Soft_drinks 1374 1256 1572 1506
Alcoholic_drinks 375 475 458 135
Confectionery 54 64 62 41
A better way to do this is fix the row names assignment at import time:
x <- read.csv(url, row.names = 1)
Q1. How many rows and columns are in your new data frame named x? What R functions could you use to answer this questions?
dim(x)
[1] 17 4
Q2. Which approach to solving the ‘row-names problem’ mentioned above do you prefer and why? Is one approach more robust than another under certain circumstances?
I prefer the second approach since it condenses the information in a more cohesive manner.
Using base R
barplot(as.matrix(x), beside=T, col=rainbow(nrow(x)))
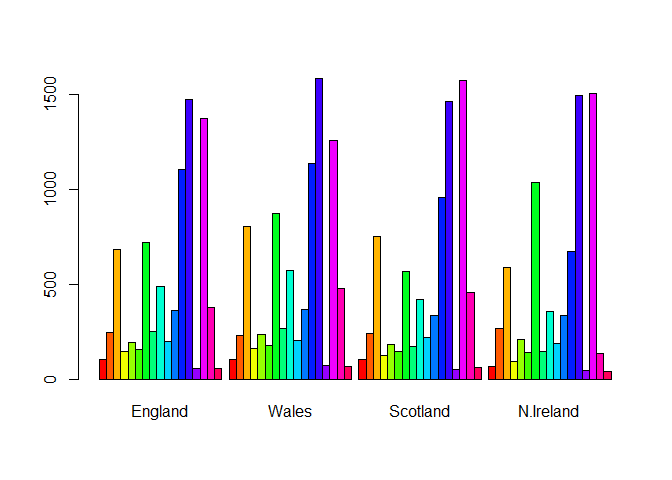
Q3: Changing what optional argument in the above barplot() function results in the following plot?
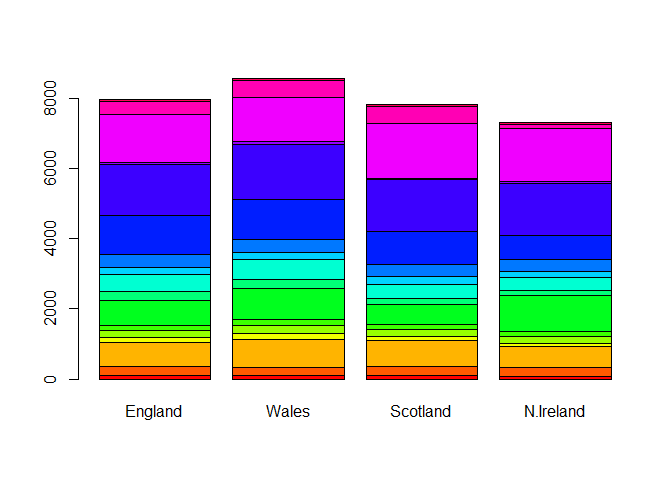
Setting beside=FALSE in your barplot() code
barplot(as.matrix(x), beside=F, col=rainbow(nrow(x)))
Pairs plots and heatmaps
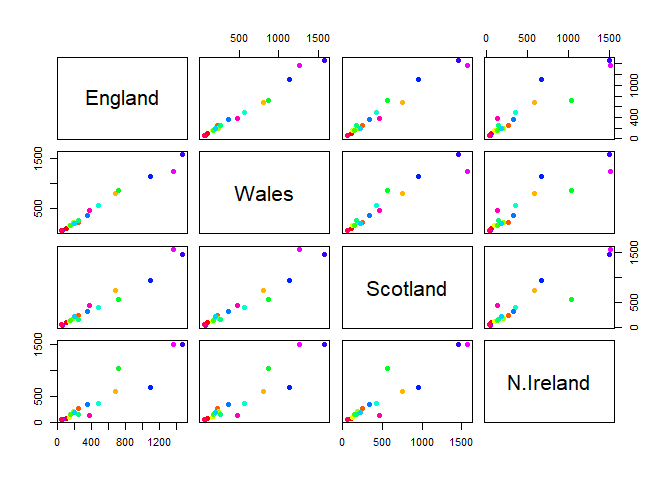
Q5: We can use the pairs() function to generate all pairwise plots for our countries. Can you make sense of the following code and resulting figure? What does it mean if a given point lies on the diagonal for a given plot?
The points are the different 17 categories such as cheese or meat. If it is on the diagona, it infers that these varying countries share similarities in the kinds of food being consumed.
pairs(x, col=rainbow(nrow(x)), pch=16)
Heatmap
We can install the pheatmap package with the install.packages()
command that we used previously. Remember that we always run this in the
console and not a code chunk in our quarto document.
library(pheatmap)
pheatmap( as.matrix(x) )
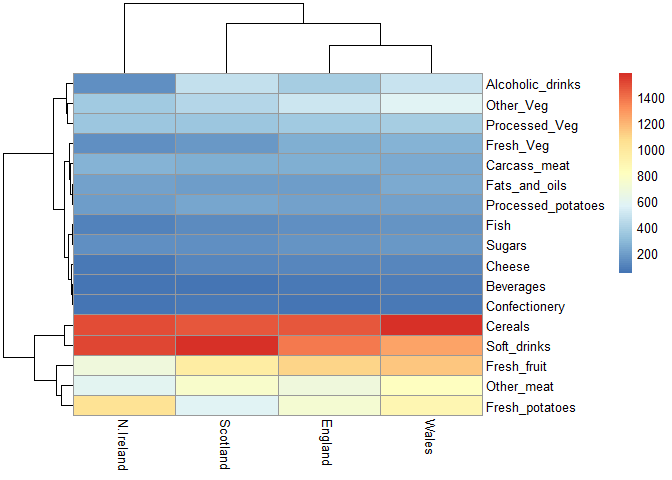
Of all these plots, really only the pairs() plot was useful. This
however took a bit of work to interpret and will not scale when I am
looking at much bigger data sets.
Q6. Based on the pairs and heatmap figures, which countries cluster together and what does this suggest about their food consumption patterns? Can you easily tell what the main differences between N. Ireland and the other countries of the UK in terms of this data-set?
Wales and England cluster together for instance, and this It is visually more appealing, however it is not the most efficient to use when comparing it to the pairs plot.
PCA to the rescue
The main function in “base R” for PCA is called prcomp()
pca <- prcomp(t(x))
summary(pca)
Importance of components:
PC1 PC2 PC3 PC4
Standard deviation 324.1502 212.7478 73.87622 3.176e-14
Proportion of Variance 0.6744 0.2905 0.03503 0.000e+00
Cumulative Proportion 0.6744 0.9650 1.00000 1.000e+00
Q. How much variance is captured in the first PC?
67.4%.
Q. How many PCs do I need ot capture at least 90% of the total variance in the dataset?
Two PCs captured 96.5% of the total variance.
Q. Plot our main PCA result. Folks can call this different things depending on their field of study e.g “PC plot”, “Ordination plot”, “Score plot”, “PC1 vs PC2 plot”…
attributes(pca)
$names
[1] "sdev" "rotation" "center" "scale" "x"
$class
[1] "prcomp"
To generate our PCA score plot we want the pca$x component of the
result object
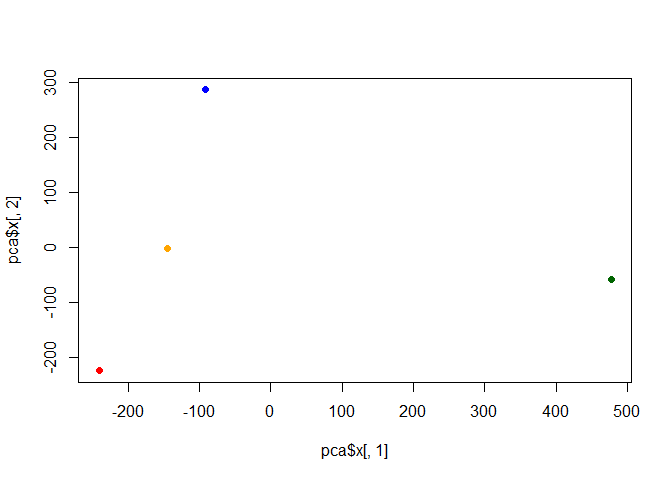
pca$x
PC1 PC2 PC3 PC4
England -144.99315 -2.532999 105.768945 -4.894696e-14
Wales -240.52915 -224.646925 -56.475555 5.700024e-13
Scotland -91.86934 286.081786 -44.415495 -7.460785e-13
N.Ireland 477.39164 -58.901862 -4.877895 2.321303e-13
my_cols <- c("orange", "red", "blue", "darkgreen")
plot(pca$x[,1], pca$x[,2], col=my_cols, pch=16)
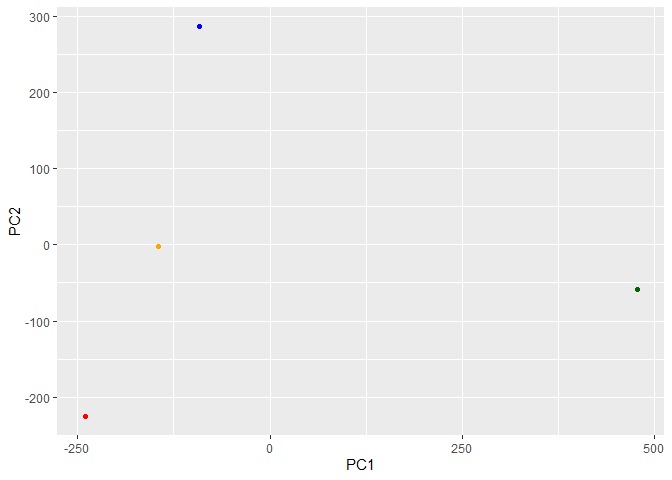
library(ggplot2)
ggplot(pca$x) + aes(PC1, PC2) + geom_point(col=my_cols)
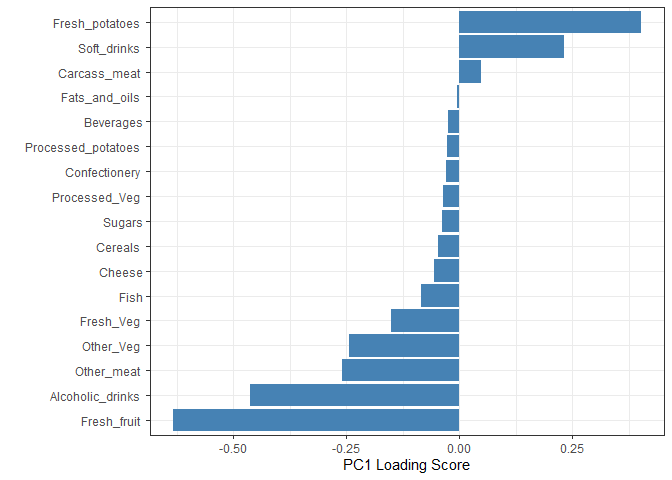
Digging deeper (variable loadings)
How do the original variables (i.e the 17 different foods) contribute to our new PCs?
## Lets focus on PC1 as it accounts for > 90% of variance
ggplot(pca$rotation) +
aes(x = PC1,
y = reorder(rownames(pca$rotation), PC1)) +
geom_col(fill = "steelblue") +
xlab("PC1 Loading Score") +
ylab("") +
theme_bw() +
theme(axis.text.y = element_text(size = 9))